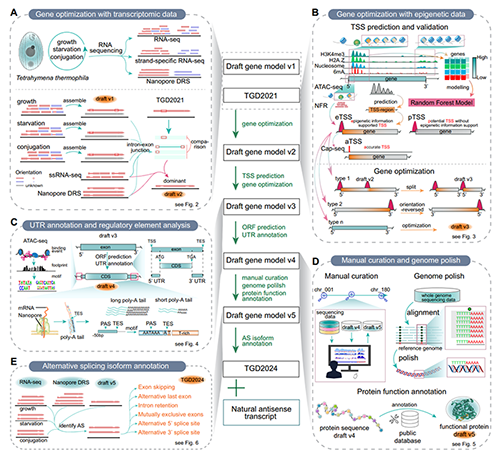
Tetrahymena thermophila is a unicellular model eukaryote, widely applied to foundational biological research. Recently, the research team led by Professor Gao Shan has optimized the genome annotation of the macronucleus of Tetrahymena thermophila by integrating large-scale transcriptomic and epigenetic data, and applying machine learning models, manual curation, and experimental validation. The research findings, titled Comprehensive genome annotation of the model ciliate Tetrahymena thermophila by in-depth epigenetic and transcriptomic profiling, were published online in Nucleic Acids Research on December 9, 2024.
The research team collected large-scale RNA-seq data of Tetrahymena spanning various cell stages (growth, starvation and conjugation). In combination with techniques such as Nanopore direct RNA sequencing (Nanopore DRS) and strand-specific RNA-seq, they carried out a comprehensive correction and re-annotation of the gene models. The team also incorporated data on epigenetic marks, including H3 lysine 4 tri-methylation (H3K4me3), histone variant H2A.Z, N6-methyldeoxyadenine (6mA) and nucleosome positioning, and applied machine learning algorithms to further optimize gene annotation. By integrating epigenomic and transcriptomic data, the team successfully predicted 24,351 transcription start sites (TSSs) and verified the accuracy of these TSSs based on Cap-seq data. Ultimately, 2,481 new genes were added to the optimized genome, and revisions including exon modifications, gene fusions, gene splits and gene inversions were completed in 23,936 gene models. In addition, the research team annotated untranslated regions (UTRs) for 26,047 genes for the first time, and identified 8,339 alternatively spliced isoforms across 5,500 genes. These optimizations have significantly improved the integrity and accuracy of the genome annotation of Tetrahymena. They not only strengthen the practical value of Tetrahymena as a genetic tool in biological research, but also offer useful references for the genome annotation of other eukaryotes.
Furthermore, the team identified 5,525 natural antisense transcripts (NATs), and found that approximately 20% of the protein-coding genes have antisense transcriptions. NATs are usually short and have low expression levels. However, during the sexual (conjugation) reproduction stage of Tetrahymena, the expression levels of NATs increase significantly. Further analysis indicated that most NATs exhibit a mutually exclusive, time-specific expression pattern with their sense protein-coding genes, and potentially regulate the transcription or translation of the sense genes through interactions. This finding offers insights into uncovering the transcriptional regulatory mechanisms in Tetrahymena.