

On October 9, 2025, a research team led by Professor Gao Shan at the Institute of Evolution and Marine Biodiversity, Ocean University of China (OUC), published a review article entitled “Sequence-independent 6mA methyltransferases for epigenetic profiling and editing” in Trends in Genetics. In this review, they systematically summarized recent advances in leveraging exogenous 6mA deposition and long-read sequencing in three major applications: chromatin landscape profiling, protein–DNA interaction mapping, and targeted epigenetic editing. For each, they highlighted their methodological advantages and discussed current challenges and prospects for optimization.
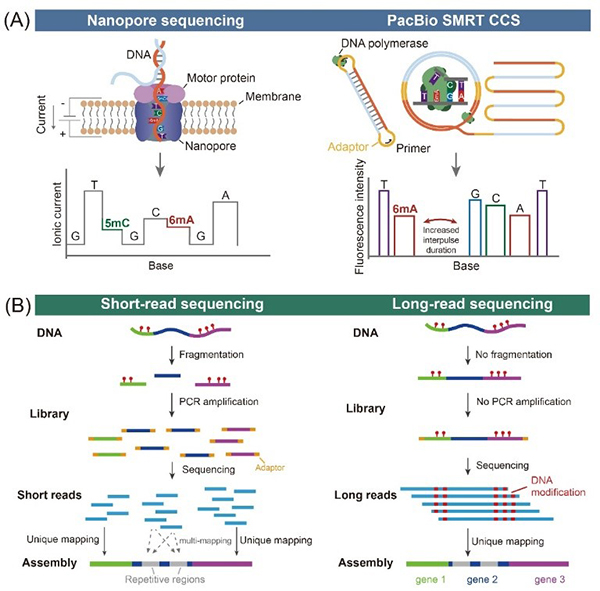
DNA N6-methyladenine (6mA), as a new epigenetic mark, is widely distributed in prokaryotes and lower eukaryotes but is relatively scarce in most higher eukaryotes, thereby providing a cleaner background for leveraging 6mA methyltransferases (MTases) to probe chromatin structure. The sequence-independent 6mA MTases, EcoGII and Hia5, due to their potent catalytic activity and broad substrate range, have become core tools in 6mA epigenetic engineering. In recent years, third-generation sequencing (TGS) technologies, especially platforms developed by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), have enabled the direct detection of DNA modifications at single-base resolution by measuring perturbations in DNA polymerase kinetics or changes in ionic current, respectively. Compared to second-generation sequencing, TGS does not require PCR amplification or DNA fragmentation, allowing for the direct detection of epigenetic marks and faithfully capturing single-molecule heterogeneity, while overcoming the mapping challenges posed by highly repetitive and structurally complex genomic regions.
By combining 6mA MTases with TGS, these approaches enable mapping of the chromatin landscape at single-molecule and single-base resolution, including nucleosome occupancy patterns, nucleosome-depleted regions, and transcription factor (TF) binding sites with high accuracy. These methods have already been applied to diverse biological systems, such as maize and human brain tissue. Nevertheless, several limitations remain, including high sequencing costs, large cell-input requirements, and constraints related to enzymatic conditions. However, as sequencing becomes more affordable and technologies such as enzyme engineering and CRISPR-based enrichment continue to advance, 6mA MTases are expected to become powerful tools for high-resolution mapping of chromatin landscape and epigenetic diagnostics.
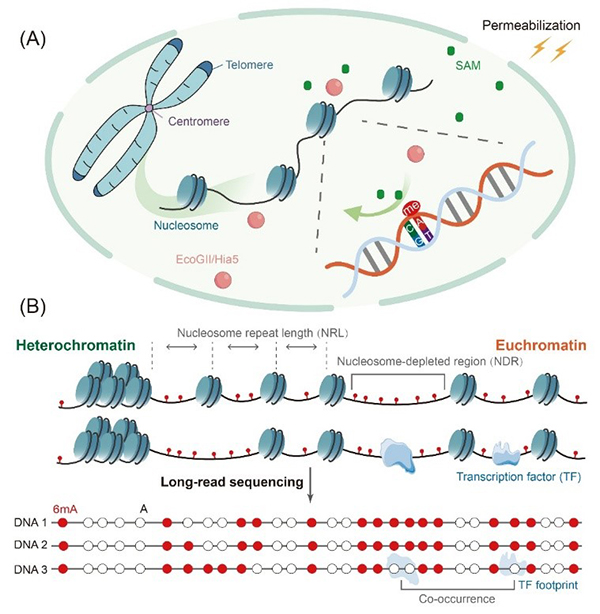
In mapping protein–DNA interactions, methods such as directed methylation with long-read sequencing (DiMeLo-seq) use antibodies to recognize a protein of interest and recruit EcoGII or Hia5 fused to protein A, thereby depositing 6mA marks specifically in the vicinity of the target protein or defined histone modifications. The resulting 6mA footprints are then read out by TGS, providing single-molecule maps of protein binding sites and revealing new protein–DNA interaction sites even within highly repetitive centromeric and subtelomeric regions. However, the stability of antibody-mediated, noncovalent bonding is limited. Future improvements, such as the introduction of nanobodies or covalent coupling strategies, are expected to enhance complex stability and thereby increase labeling efficiency and the reliability of site-specific readout.
In targeted epigenetic editing, 6mA MTases such as METTL4 can be fused to dCas9 to achieve site-specific installation of 6mA marks in various cell types. By recruiting 6mA reader proteins and TFs, these tools enable precise control of gene expression at defined genomic loci. Notably, because EcoGII and Hia5 exhibit minimal sequence constraints, they are attractive scaffolds for constructing 6mA-based editing tools; however, their catalytic activity needs to be optimized through vector design and orthomutation, thereby reducing off-target effects.





